Bulked-segregant analysis簡稱BSA,也叫(jiào)混合分組分析,是一種(zhǒng)利用樣本混池的建庫方式對(duì)動植物的極端性狀進(jìn)行QTL定位的一種(zhǒng)方法。這(zhè)種(zhǒng)方法快速便捷,在親本群體中選擇表型極端的個體構建表型極端的子代群體,并對(duì)極端性狀的子代群體進(jìn)行混池測序,從而迅速定位到與目标基因具有緊密連鎖的分子标記區域,進(jìn)一步挖掘重要的功能(néng)基因。Graded-seq是升級版的BSA,可對(duì)多個混池DNA進(jìn)行分析。
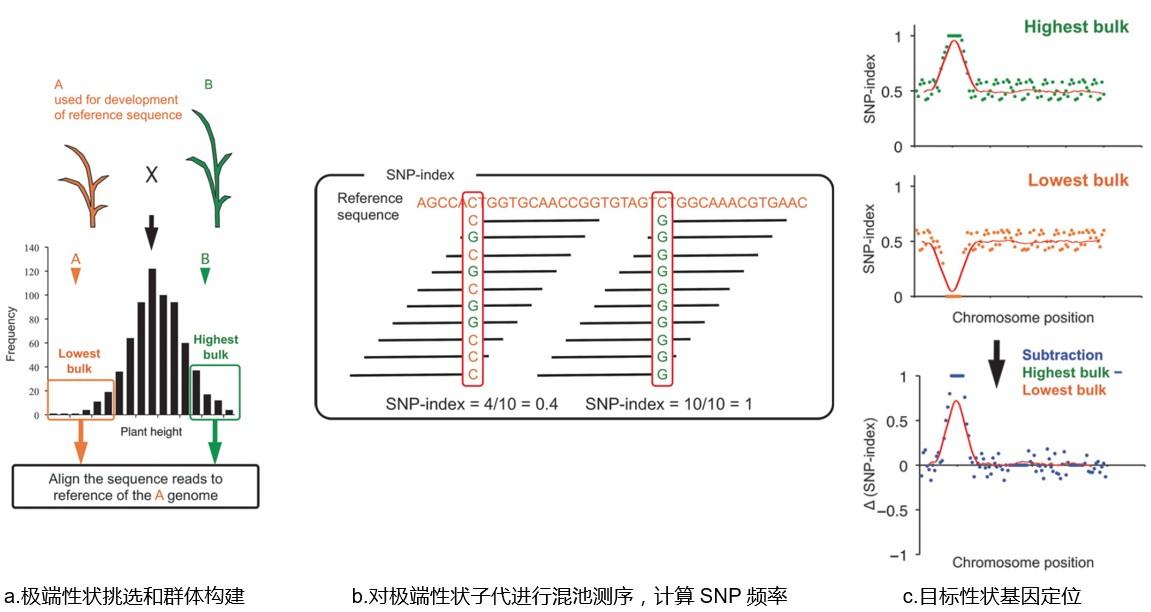

取樣類型:具有重要極端性狀的親本,子代群體包括暫時(shí)分離群體F1、F2和永久分離群體RIL、NIL和DH
混池規模:子代數量最少30+30個起(qǐ),推薦數量50+50個起(qǐ)。必須符合極端性狀的要求。
測序深度:親本測序深度最低20x起(qǐ),推薦深度30x起(qǐ)。平均每個子代1x起(qǐ),推薦2x起(qǐ)。
Q1:混池數量有要求嗎?
A:子代混池數量最少30個起(qǐ),建議至少50個。理論上混池數量越多,性狀定位越好(hǎo)。如果極端性狀的樣本數量不多不建議強行湊樣,最低至少也要達到20個左右。兩(liǎng)種(zhǒng)極端性狀的混池數量可以不一緻。
Q2:沒(méi)有親本,隻有RIL群體或者是F2群體,可以做BSA嗎?
A:可以做。聯川生物會(huì)采用ED分析法進(jìn)行分析。但是定位區間内的候選基因會(huì)偏多。
A:建議老師親本測序深度至少要10x-20x以上,推薦30x以上。子代混池,平均每個子代至少要1x以上,推薦2x以上。理論上測序深度越深,标記的準确性會(huì)越強。
A:是的。Mutmap僅限于EMS等化學(xué)誘變劑引起(qǐ)的點突變。
A:可以的。如果沒(méi)有參考基因組可以做BSA但是後(hòu)期數據關聯性較差,不太推薦做BSA,做簡化基因組遺傳圖譜較爲合适。另外針對(duì)一些沒(méi)有參考基因組但是基因組雜合度較高的物種(zhǒng)如牡丹,推薦做BSR混池。
A:我們不建議老師使用動物樣本做BSA混池,這(zhè)裡(lǐ)的動物不包括魚、昆蟲等物種(zhǒng)。但是我們一般會(huì)優先推薦植物,二倍體和多倍體都(dōu)是可以做混池的。
快捷高效的BSA混池測序鑒定水稻抗稻瘟病QTL
研究背景
使用傳統的育種(zhǒng)方法鑒定某個重要的農藝性狀存在費時(shí)費力等缺點,采用BSA策略可以通過(guò)對(duì)性狀差異較大的親本進(jìn)行雜交,快速構建作圖群體如F2、RIL等。在亞洲水稻是一種(zhǒng)十分重要的糧食作物,鑒定重要農藝性狀的相關基因迫在眉睫。
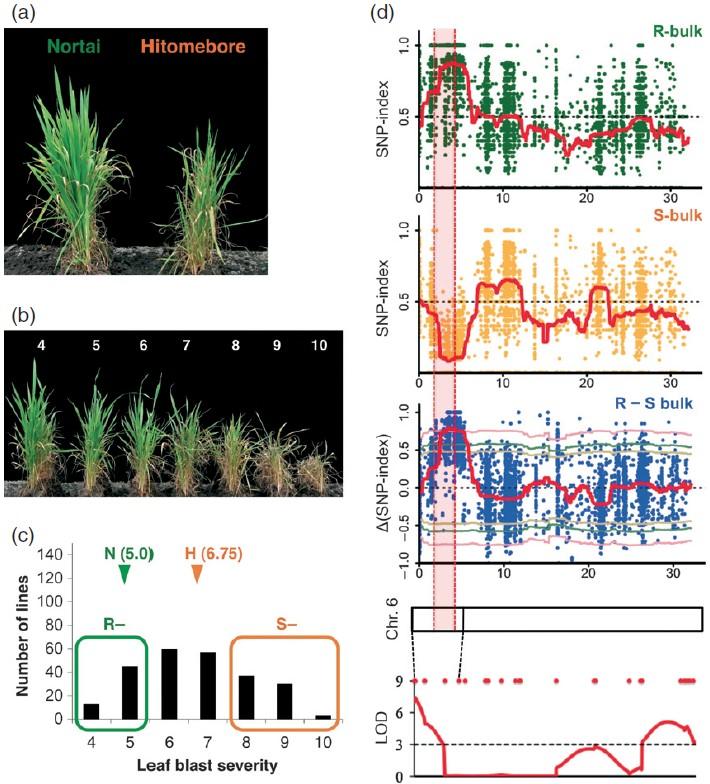
來自日本的Terauchi教授和他的同事(shì)們,利用抗稻瘟病的水稻品系Nortai和感病水稻品系Hitomebore(圖a)雜交後(hòu)得到F2群體,之後(hòu)連續自交得到總計241株水稻(RIL群體)。之後(hòu)按照不同的抗病能(néng)力對(duì)RIL群體進(jìn)行分級(圖b),將(jiāng)抗病能(néng)力最強的20株水稻和抗病能(néng)力最弱的20株水稻分别構建2個混池(圖c)。
SNP-index和基因組位置關系圖可以看出,抗病混池(R-bulk)和感病混池(S-bulk)的SNP-index在6号染色體上差異較大。△SNP-index(R-S bulk)可以看出,染色體上絕大部分區域的△SNP-index都(dōu)接近于0,而2.39-4.39Mb這(zhè)段區域的△SNP-index都(dōu)大于0.79且p<0.01(圖d)。
接下來作者使用最傳統的遺傳圖譜法,對(duì)所有241株水稻進(jìn)行QTL定位,發(fā)現0-4.9Mb内LOD值最高,去交集後(hòu)抗稻瘟病的區域進(jìn)行快速定位。
Takagi H, Abe A, Yoshida K, et al. QTL-seq: rapid mapping of quantitative trait loci in rice by whole genome resequencing of DNA from two bulked populations. Plant J, 2013, 74(1):174.
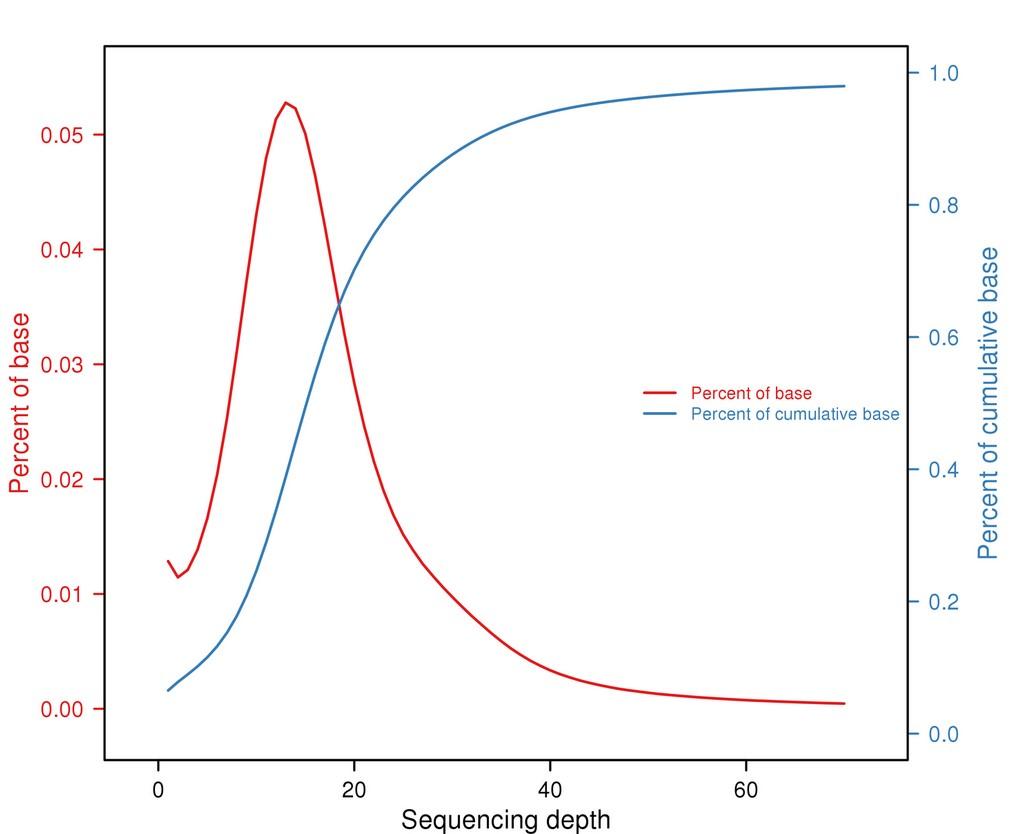
Reads比對(duì)到參考基因組後(hòu),可以計算堿基的覆蓋到基因組上的比例。參考基因組上被reads覆蓋到的堿基數占基因組的百分比稱爲基因組覆蓋度;堿基上覆蓋的reads數爲覆蓋深度。
使用GATK進(jìn)行SNP Calling和Indel Calling,接下來使用ANNOVAR對(duì)SNP和Indel結果進(jìn)行注釋。
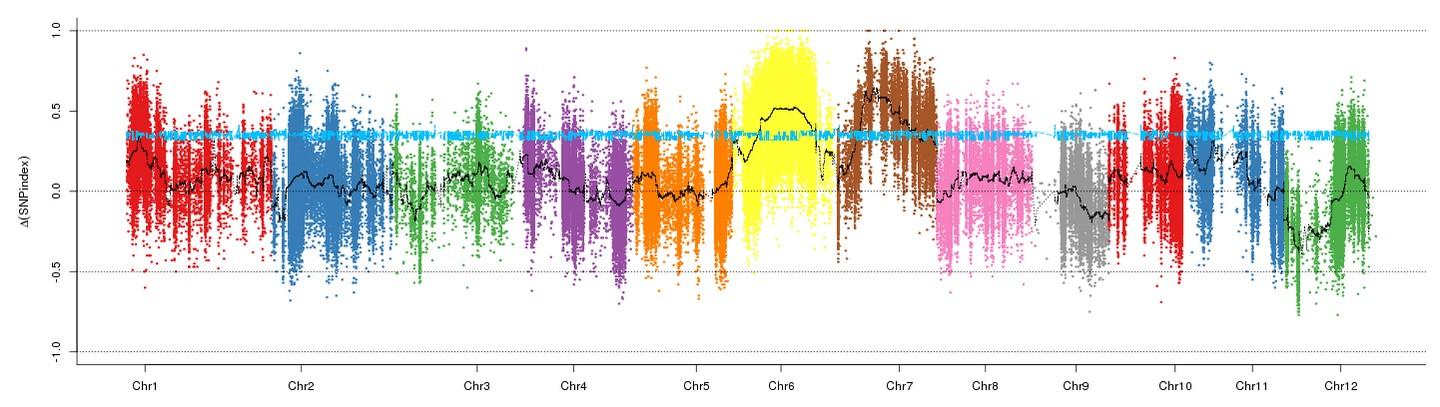
根據親本和混池的SNP及Indel标記,利用SNP-Index和ED分析方法同時(shí)進(jìn)行标記與性狀的關聯分析,獲得與目标性狀相關的基因。
使用聯川生物自主開(kāi)發(fā)的富集分析程序,對(duì)目标性狀區域内的候選基因進(jìn)行功能(néng)富集分析。