背景簡介
細菌全基因組具有相對(duì)較小、重複序列較高、易于突變等特點,通過(guò)全基因組測序,可以對(duì)細菌基因組進(jìn)行測序、拼接、組裝,獲得完整細菌基因信息。細菌de novo測序已取代傳統方法成(chéng)爲研究細菌進(jìn)化遺傳機制、關鍵功能(néng)基因的重要工具。目前,細菌全基因組測序利用三代測序結合二代測序的方法可以得到0 Gap的完整的基因組序列;對(duì)其進(jìn)行功能(néng)基因注釋以及個性化分析,全面(miàn)解析細菌生物學(xué)意義。
技術優勢
實驗安排靈活、服務周期短、速度快
測序結果準确性更高、成(chéng)本更低
豐富的項目經(jīng)驗與專業的生信分析團隊,提供最全面(miàn)準确的信息分析
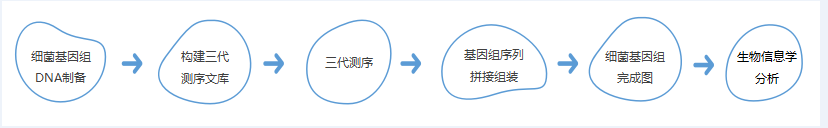
技術路線
分析内容

樣本類型
DNA送樣:
DNA濃度 ≥20ng/μl(Qubit),DNA質量≥15ug(Qubit)DNA電泳條帶單一,無明顯降解。
菌體送樣:
收集生長(cháng)對(duì)數期菌體,收集離心菌體數3×1010個,于無菌離心管中,液氮速凍,幹冰運輸
Q1:爲何完成(chéng)圖選擇三代測序平台?
A:受測序片段長(cháng)度的限制,細菌基因組序列通常需要利用軟件算法將(jiāng)大量測序片段拼接起(qǐ)來,而細菌基因組中重複序列的存在,則會(huì)大大增加拼接的複雜度。細菌重複序列的大小從幾百bp到7 Kb不等,細菌框架圖的插入片段,隻能(néng)解決少量的重複片段問題,因此組裝結果更加碎片化;細菌精細圖采用了6 Kb大片段文庫,可以跨過(guò)絕大部分重複序列,并將(jiāng)結果Scaffold控制在30條以内;而三代測序采用了10 Kb文庫,平均讀長(cháng)也達到10 Kb以上,由于序列夠長(cháng),避免了細菌基因組中重複序列的影響,因此能(néng)夠獲得0 gap的完整組裝結果。
Q2:對(duì)于細菌基因組測序,三代和二代測序相比有何優勢?
A:三代測序相比二代測序而言,其優勢在于讀長(cháng)長(cháng),GC含量影響小,而劣勢是測序成(chéng)本偏高。對(duì)于細菌基因組測序來說(shuō),三代測序的長(cháng)讀長(cháng)可以解決細菌中的重複序列問題,也避免了異常GC菌株的測序不均勻問題。由于細菌基因組較小,需要的測序量不大,對(duì)于較爲精細的細菌完成(chéng)圖來說(shuō),三代成(chéng)本甚至低于二代結合一代的策略。目前爲止,在需要組裝完整性較低的細菌框架圖層面(miàn),二代測序仍能(néng)保持一定成(chéng)本優勢。随著(zhe)三代測序通量提升和成(chéng)本降低,未來三代測序有望在細菌基因組領域獲得更廣泛的應用。
Q3:細菌基因組中如何預測核糖體rDNA基因?
A:預測細菌基因組中的核糖體rDNA基因,通常有兩(liǎng)種(zhǒng)方法:一是通過(guò)rDNA序列結構特征進(jìn)行de novo 預測,二是利用近緣rDNA序列進(jìn)行同源預測。其中前者預測更準确,但是需要組裝結果中具備完整的rDNA結構。在框架圖和部分精細圖組裝結果中,可能(néng)有rDNA區域組裝不完整,分布于多條scaffold中的情況,會(huì)導緻de novo 測序方法rDNA預測不到的情況。如果想要獲得更完整的預測結果,可以預先提供近緣rDNA序列,使用同源預測方法,以改善預測效果。
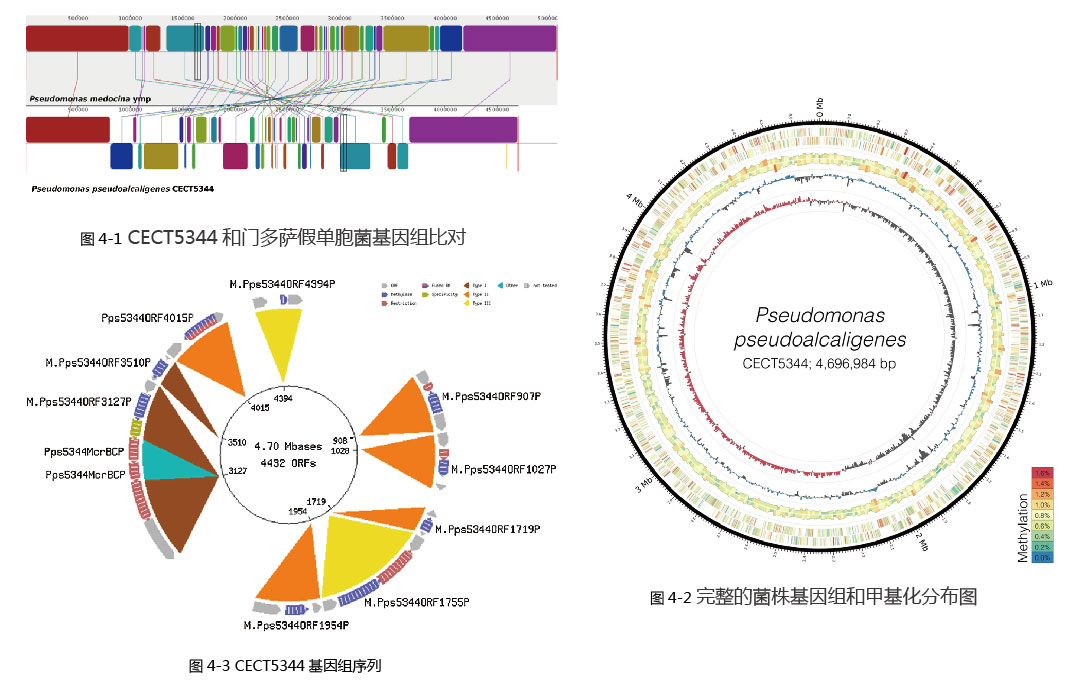
通過(guò)單分子實時(shí)測序解析可降解氰化物的産堿假單胞菌CECT5344基因組完成(chéng)圖和甲基化情況
研究背景
産堿假單胞杆菌CECT5344在耐受氰化物的同時(shí),還(hái)可以在堿性條件下利用氰化物和氰基衍生物作爲氮源,極可能(néng)作爲含氰液體廢液污染的生境的生物修複菌。之前已經(jīng)有該菌株的基因組序列信息,現在采用單分子實時(shí)監測序列技術(SMAT)對(duì)其基因組進(jìn)行重測序,得到由GC含量爲62.34 %,長(cháng)4696,984 bp的完整基因組序列。重測序得到的基因組補充了原來基因組中遺漏的部分片段信息,這(zhè)些遺漏的片段多爲轉座因子,此外還(hái)發(fā)現了預測在亞砜還(hái)原中起(qǐ)作用的5個基因。CECT5344的基因序列與門多薩假單胞菌高度同源,兩(liǎng)者約有70%的基因是相同的。與門多薩假單胞菌不同,CECT5344中并沒(méi)有發(fā)現推斷的緻病性基因。CECT5344擁有氰水解酶和汞抗性蛋白的獨特基因,這(zhè)些對(duì)被氰基和汞化合物污染的環境緩解尤爲重要。通過(guò)SMAT測序還(hái)可以得到菌株的m6A類型的甲基化信息。菌株CECT5344的完整基因組序列爲生物學(xué)遺傳特征的研究提供了基礎。
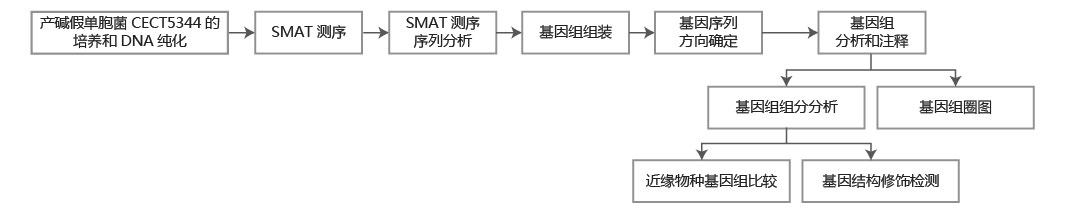
方法流程
研究結果
對(duì)CECT5344和門多薩假單胞菌基因組比對(duì),對(duì)兩(liǎng)者的基因結構關系進(jìn)行了研究,圖 4-1中用線連接的色塊表示兩(liǎng)個基因組的同源區域,最下方的色塊代表門多薩假單胞菌基因組中與CECT5344基因組反向(xiàng)的區段。
圖 4-2爲完整的菌株基因組和甲基化堿基分布圖,基因組由4,696,984個堿基對(duì)和4436個預測的編碼序列組成(chéng)。圓圈從内到外分别代表:GC偏斜、GC含量、50 kb窗口分析的全基因組甲基化、每個基因的鏈特異性甲基化、每百萬堿基對(duì)甲基化的量。
使用REBASE數據庫尋找到CECT5344基因組中編碼甲基轉移酶的基因,共鑒定預測了9個限制/修飾基因的基因座。圖 4-3中顔色代碼表示不同的限制/修飾類型,藍色的爲甲基化酶,紅色爲限制性酶。最内層爲菌株的基因組完整圖和開(kāi)放閱讀框的數目。
參考文獻
Daniel Wibberga, Andreas Bremgesb, Tanja Dammann-Kalinowskia,et al.Finished genome sequence and methylome of the cyanide-degradingPseudomonas pseudoalcaligenes strain CECT5344 as resolved bysingle-molecule real-time-sequencing.Journal of Biotechnology, 2016, 232:61-68.
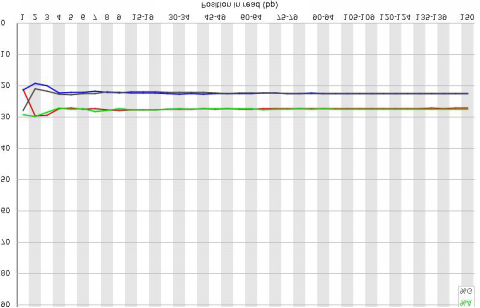
原始數據堿基組成(chéng)分布例圖橫坐标是reads 堿基坐标,縱坐标是所有reads 的A、C、 G、T、N 堿基分别占的百分比。每個位置上,A、C、G、 T在開(kāi)始有所波動,後(hòu)面(miàn)會(huì)趨于穩定。一般情況下A 與T 相 等,C與G相等,各堿基所占百分比會(huì)因物種(zhǒng)差異而不同。 基因組項目中,建庫比較均勻的情況下,代表不同堿基的 四種(zhǒng)顔色的分界線應該波動極小。
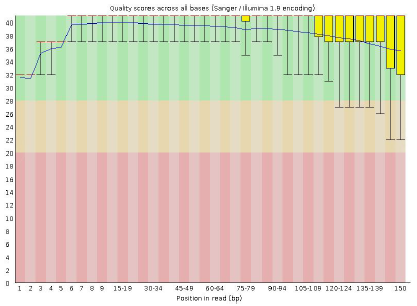
原始數據堿基質量分布例圖橫坐标是reads 堿基坐标,縱坐标是reads 的堿基質量(SolexaScale: 40=Highest, - 15=Lowest),圖中垂直紅線”Ⅰ”指定的範圍是所有reads 堿基的綜合質量,紅色垂直方塊是質量的四分位值範圍,加黑粗線是質量值的中位數。
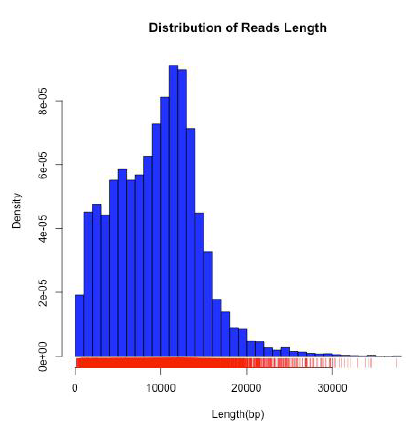
單分子Clean 數據序列的長(cháng)度分布統計圖橫坐标爲測序reads 的長(cháng)度,縱坐标爲不同長(cháng)度reads的數目,從上圖中可以看出,本次測序獲得的reads的長(cháng)度大小主要集中分布在5000-15000bp,測序質量較高。