遺傳圖譜也叫(jiào)連鎖遺傳圖譜(Genetic linkage map),是指基因或DNA分子标記在染色體水平上的相對(duì)位置與遺傳距離,通常以基因或DNA片段在染色體交換過(guò)程中的重組頻率來描述,單位爲厘摩爾(cM)。例如兩(liǎng)個基因或兩(liǎng)個分子标記之間的遺傳距離爲0.8cM表示減數分裂時(shí)的重組頻率爲0.8%。這(zhè)裡(lǐ)的遺傳距離指的是相對(duì)距離而不是染色體上的物理距離。兩(liǎng)者的遺傳距離越近,發(fā)生重組的概率就(jiù)會(huì)越低,反之亦然。 數量性狀是指表型呈現連續變化的性狀,如株高抗病能(néng)力等容易受到多種(zhǒng)因素的影響。控制數量性狀的基因在基因組上的位置被稱作數量性狀基因座(Quantitative Trait Locus, QTL)。 尋找QTL在染色體上的位置并估計其遺傳效應,稱作QTL作圖(QTL mapping)。構建圖譜需要有大量的分子标記作爲輔助,傳統的分子标記包括RFLP、SSR等。但是用這(zhè)些标記去定位區間存在标記密度過(guò)低,構建費時(shí)費力等問題,逐漸被SNP标記給替代。利用全基因組重測序或簡化基因組測序的方法得到高密度的SNP标記定位QTL已經(jīng)成(chéng)爲主流。
應用領域
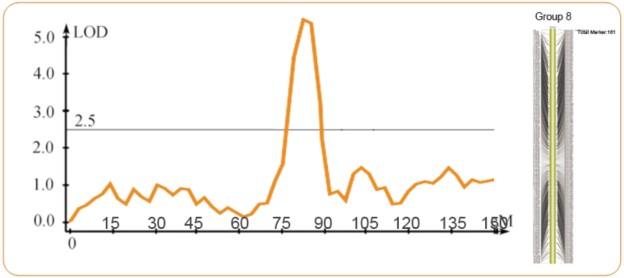
QTL定位:通過(guò)表型性狀與标記間的關聯分析來确定各個數量性狀位點在染色體上的位置、效應及各個QTL間的 相互作用。
輔助基因組組裝:通過(guò)帶有序列信息的遺傳圖譜,將(jiāng)組裝的scaffold進(jìn)行進(jìn)一步的鏈接,并定位到染色體上,以獲得基因組精細圖譜。
分子标記輔助育種(zhǒng):獲得與性狀緊密連鎖的标記,根據标記序列設計引物,直接應用于群體中的性狀篩查。

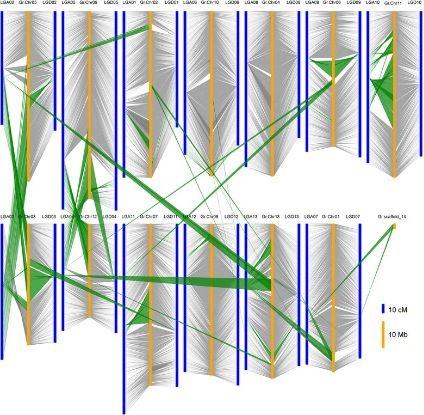
比較基因組學(xué)研究:利用高密度遺傳圖譜上标記的宏觀共線性,揭示不同物種(zhǒng)間染色體或片段上的共線性,分析相關物種(zhǒng)基因組結構和進(jìn)化曆程。
方案設計
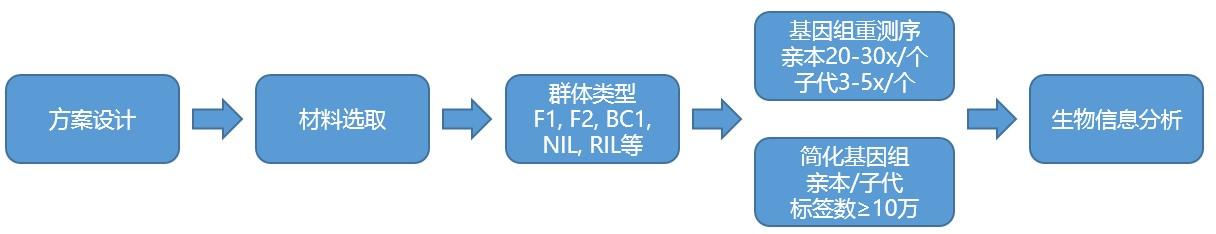
分析内容
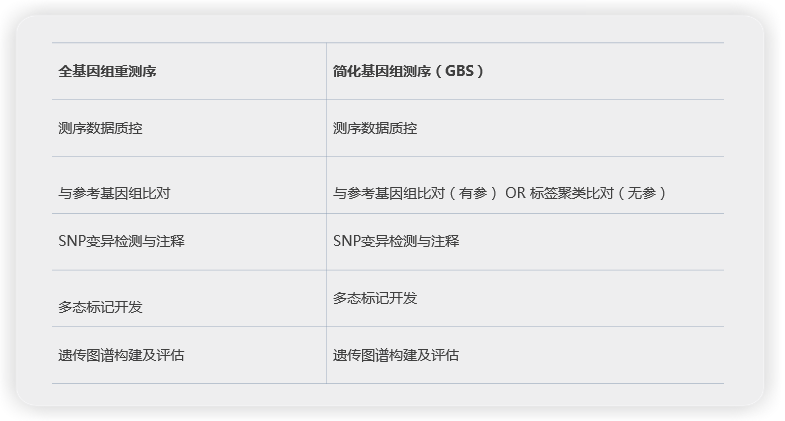
樣本類型和測序方案
取樣類型:具有重要性狀的親本,雜交或自交能(néng)産生後(hòu)代。
子代群體包括暫時(shí)分離群體F1、F2和永久分離群體RIL、NIL等
作圖群體:父本母本各一個,子代數量150個起(qǐ)。随機挑選作圖群體。
測序深度:親本20-30X,子代3-5X
A:我們一般推薦老師的樣本數≥200,至少150個起(qǐ)。和BSA混池選樣策略完全相反,即BSA選取的是極端性狀做混池,而遺傳圖譜盡量做到随機取樣,而不是刻意挑選。
A:首先要符合親本雜交或自交後(hòu)具有可育性,然後(hòu)根據親本的雜合和純合度選擇分離群體。通常情況下親本純合度較高的物種(zhǒng)如水稻等,分離群體可選用F2、BC、RIL、DH、NAM群體等。而雜合度較高的親本(純合度<50%)如林木、果樹、水生魚類、昆蟲等一般采用F1作爲分離群體。
A:對(duì)于親本,我們推薦測序深度≥20X。子代推薦深度3-5X。
A: 同源多倍體物種(zhǒng)遺傳圖譜目前可以當做二倍體來做,異源多倍體例如四倍體陸地棉等作物可以當做兩(liǎng)個二倍體作物來構建遺傳圖譜。
A:遺傳圖譜用途廣泛,包括QTL定位、輔助基因組組裝、分子标記輔助育種(zhǒng)和比較基因組學(xué)研究等。
A:事(shì)實上表型數據中的随機誤差大緻符合正态分布即可,無需嚴格要求表型數據滿足正态分布。但是這(zhè)裡(lǐ)需要再次強調的是,老師在選擇作圖群體時(shí),取樣盡量要均勻随機而不是刻意選擇自己喜歡的性狀。
A:包含偏分離不顯著的标記,顯著偏分離的标記會(huì)被過(guò)濾掉。偏分離是自然界非常普遍存在的現象,并被認爲是生物進(jìn)化的動力之一。産生偏分離的原因主要有兩(liǎng)個方面(miàn):配子選擇和合子選擇,其中配子選擇主要包括花粉緻死、花粉管競争和選擇性受精。除了上述原因以外,環境因素、非同源重組、基因轉換、轉座因子、轉基因沉默、體外孤雄生殖過(guò)程中的選擇壓、非整倍體或不穩定易位造成(chéng)的染色體不穩定等都(dōu)有可能(néng)是導緻偏分離。
材料:
親本: Magellan ×PI 438489B
子代:RIL群體(246個子代個體)
測序
親本測序深度13.5X,子代 0.19X
性狀
抗豆根受線蟲病
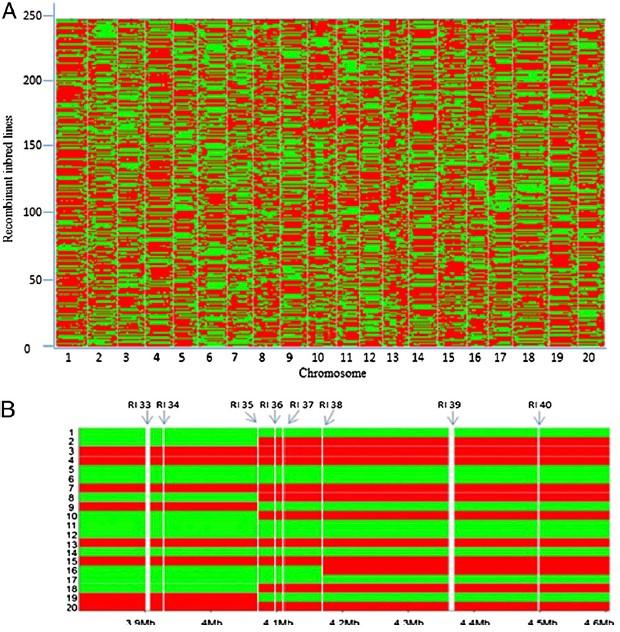
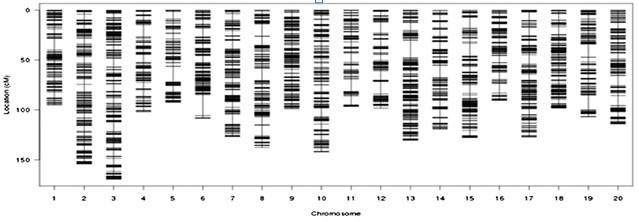
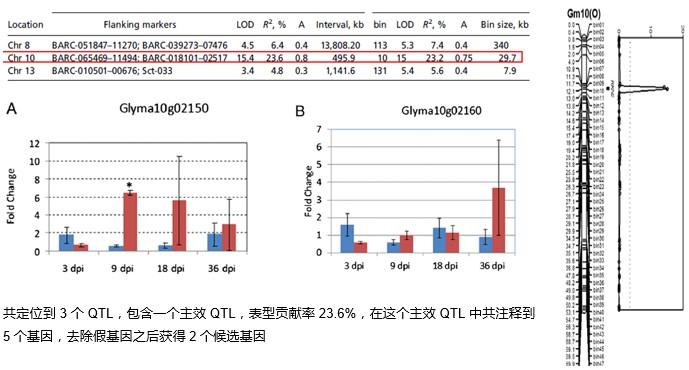
在群體足夠大的情況下,采用重測序的方法測序定位效果會(huì)更加精細;尋找非同義突變位點,縮小候選區域;一步到位,直接獲得候選區域序列信息。
重測序可以高效開(kāi)發(fā)雙親之間的SSR,InDel标記同時(shí)檢測SV,CNV變異信息;SSR,InDel标記檢測操作相對(duì)于CAPS,dCAPS标記更加容易,一般實驗室就(jiù)能(néng)順利完成(chéng)。
可以與參考基因組和近緣物種(zhǒng)進(jìn)行共線性分析,尤其是與近緣種(zhǒng)進(jìn)行共線性分析時(shí)可以挑出同源片段,深入研究這(zhè)些保守片段的分子結構和特點。
利用親本的高深度序列信息(30X)可以完善參考基因組序列信息,輔助和糾錯基因組裝工作。
 Xu, X., et al. (2013). Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. PNAS, 110(33), 13469-74.
Xu, X., et al. (2013). Pinpointing genes underlying the quantitative trait loci for root-knot nematode resistance in palaeopolyploid soybean by whole genome resequencing. PNAS, 110(33), 13469-74.
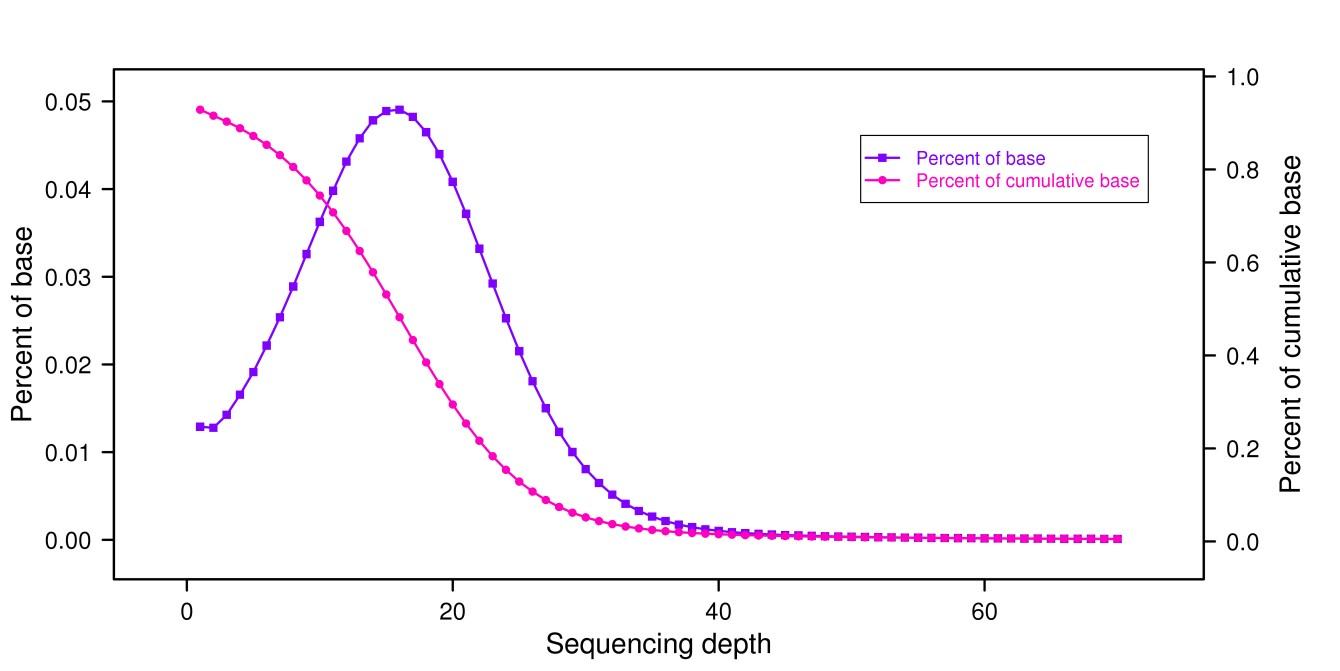
測序深度分布圖
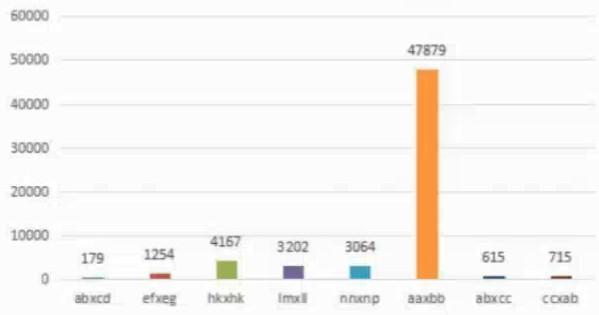
多态标記開(kāi)發(fā)
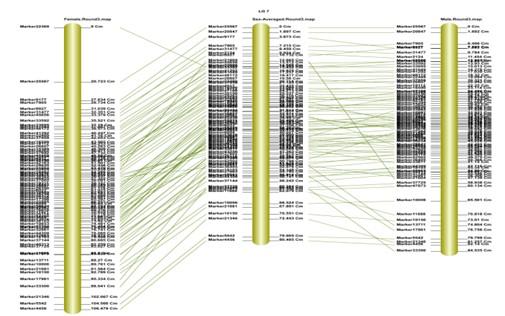
遺傳譜圖共顯性評估
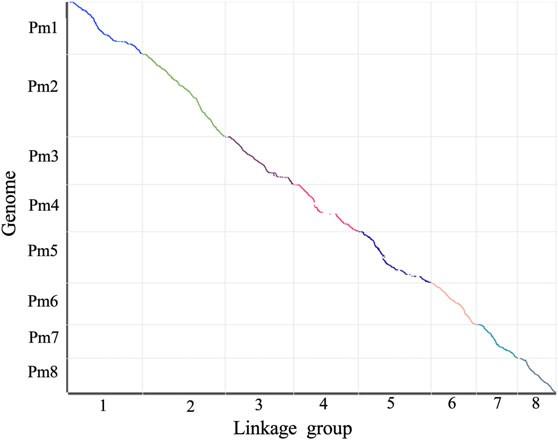
圖譜質量評估
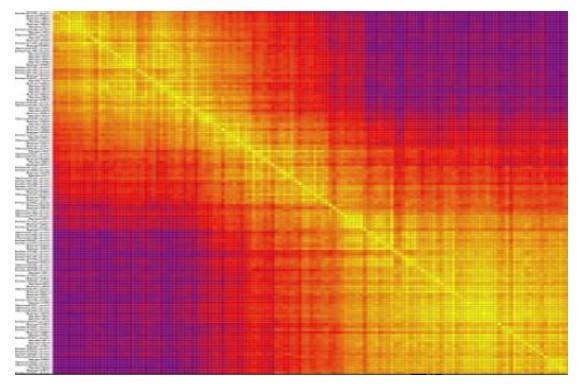
共線性評估

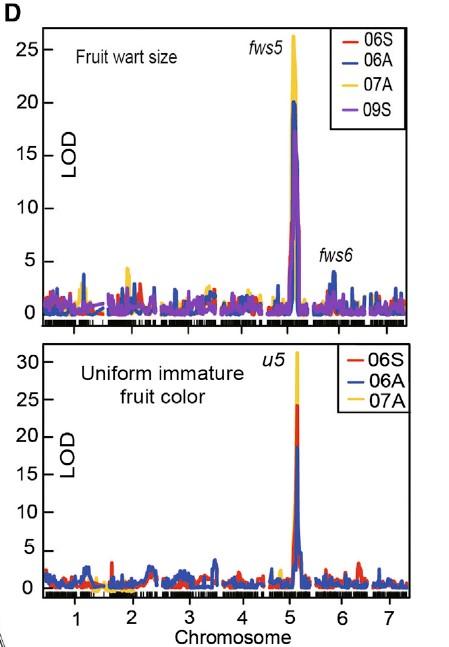
QTL定位結果
遺傳圖譜構建
